Atlantoaxial (AAI) and craniocervical instability (CCI) are two potentially sinister diagnoses that cause damage to the segmental neurovascular structures due to overmobility of the upper cervical spine. This, usually due to trauma, but can also occur gradually due to certain autoimmune disorders such as rheumatoid arthritis, gross congenital hypermobility (such as Ehler Danlos syndrome or Marfan syndrome), or certain congenital syndromes such as Down’s syndrome (Yang et al. 2014).
Atlantoaxial instability will generally imply axial hypermobility of the atlantoaxial joint itself, which when symptomatic will result in Bow hunter’s syndrome (positional compression or damage to the vertebral arteries) or Cock Robin syndrome (positional facetal dislocation without reduction). Luxation of the atlantoaxial joints, ie., luxation that surpasses what is seen in Cock Robin syndrome, can also occur with traumatic and gross ligamentous rupture. The term AAI can also be used in cases of transverse ligament rupture, in which the odontoid process (the axis of the C2) may, especially if there is also damage to the tectorial membrane, dislocate dorsally and compress the brainstem. Brainstem compression, when symptomatic, will usually cause quadriparesis along with phrenic nerve palsy. Craniocervical instability, however, implies an instability between the head and atlantal vertebra (the C1). Traumatic ligamentous ruptures or gradual deterioration of joint stability may cause basilar invagination, which is a degenerative process causing the odontoid process to graduall migrate into the head via the foramen magnum. This can also damage the brainstem and produce symptoms similar to what is described above.
Traditional cases of atlantoaxial instability and craniocervical instability require obvious imaging findings with strong clinical correlation, and, when its criteria are met, are certainly treated (operated) in any skilled and compatible neurosurgical ward. For example, if the patient blacks out every time she turns her head to the left, a followup dynamic catheter angiography could be done, and may demonstrate high-grade stenosis of the vertebral artery when turning to the left. This is Bow hunter’s syndrome, and may be caused by legitimate atlantoaxial instability. Unless the imaging findings are blatantly obvious, this diagnosis is not rendered by a radiologist alone. Strong evidence of clinical correlation must be present from a clinician that is familiar with the signs and triggers in upper cervical instability-cases.
There is a growing trend, however, within the (or, at least, certain) alternative medical communities, where patients with normal or virtually normal imaging, and with the absence of clinical triggers that would suggest atlantoaxial or craniocervical instability, still end up diagnosed with these relatively sinister diagnoses. Often, by radiologist alone, based on sparsome imaging findings (eg., alar ligament T2 FLAIR hyperintensity or mild to moderate lateral facetal overhangs) and a lacking compatible clinical workup. Hopefully, this is the result of ignorance combined with poor clinical workup skills (incompetence) and not mere greed and malevolence. Regardless, be it rooted in benevolent or malevolent intention, this does not change the fact that pursuing the diagnosis and especially its related treatment (conservative or surgical strategies) are extremely expensive and potentially dangerous as well. Clearly, the expenses involved, including the health risks, may be well worth it if the diagnosis is correct and the patient has legitimate CCI or AAI with strong clinical and radiological evidence. However, can we say the same if there is major guesswork involved in the rendering of the diagnosis?
This article will take a critical look at these diagnoses and elaborate upon the factual structural risks that are seen in atlantoaxial- and craniocervical instabilities, as well as their expected realistic symptoms and triggers. The triggers would be especially relevant, seeing as various symptoms can heavily overlap between hundreds if not thousands of diagnoses.
Implicit clinical correlation in legitimate AAI or CCI
It is crucial to understand that the general minor instabilities involved in AAI and CCI are not the cause of symptoms. A common but severely ignorant misunderstanding that some clinicians make (the patient cannot be blamed for thinking like this, but the clinician should set it straight), is the notion that mild to moderate ligamentous instabilities makes the neck (or the whole body for that matter) tense up to protect against the ligamentous instability, even though there are minimal or no clear MRI findings to support this notion, and that this somehow causes all of the patient’s symptoms. Some – top offenders – may suggest full craniocervical fusion, ie. fusion from the head, all the way down to the T1 or T2 vertebrae, even though there may be zero evidence for major neurovascular conflict. In reality, in legitimate cases of atlantoaxial or craniocervical instability, the instability may cause a potentially dangerous neurovascular conflict, as mentioned initially, where the brainstem or vertebral arteries can get damaged. Surgical options, sometimes including relevant-level fusion, may be warranted in these circumstances. But we must see adequate imaging as well as adequate clinical fulfillment of diagnostic criteria to render these diagnoses; it is not enough to feel neck clunking, upper cervical pain, weakness in the neck or wobbleheaded. There can be, and are indeed many more potential explanations for these symptoms than just AAI and CCI. Why would you jump to the worst possible explanation, and especially when lacking apt evidence?
Many of these patients who have been misdiagnosed with AAI or CCI may feel neck “wobbliness”, heaviheaded, neck weakness, and clicking or clunking in the neck upon movement, often along with upper neck pain. This may cause the patient to become afraid and to google their symptoms, which in and by itself is reasonable enough. The problem begins when certain nonsensical articles about CCI and AAI, that do not properly explain relevant clinical correlation nor imaging requirements, but rather, just lists a set of associated symptoms, finds favor in the patient. The patient may seek out their GP or a local neurosurgeon who will, usually, and usually rightfully so, dismiss these claims, as the patient’s imaging is normal and also lack neurological signs that would fit with neurovascular compromise. The problem, in the patient’s eyes, may be a lacking reasonable counter-argument and counter-diagnosis that would explain his or her symptoms, which then prompts the patient to seek out alternative health care. This can be a blessing if one proceeds to be properly diagnosed based on objective criteria, but often extremely expensive and also dangerous, if not. How is one supposed to know, if no one knows what you have in the first place? I am not saying it is easy.
Common arguments for treatment may be claims that, although the MRI and even upright MRIs are normal, their own DMX scan is positive, or that the MRI, which was deemed normal by the local hospital, in reality shows signs of ruptured ligaments and that this fits with the patient’s symptoms. Now, it is true that specialty diagnoses can be missed by local generalists. But, if a specialist points something out that is not conventionally considered, he should either 1. make sure to emphasize the notion that it is a subtle finding with unsure actual clinical applicability or 2. make sure to prove his points through objective findings. Unfortunately, and this is a big problem, even if the clinician makes up a nonsencial argument, or if they offer an evidence based objective opinion, the patient will rarely have the necessary medical knowledge to discern between the two, and will, ultimately, guide their decisions by faith [or lack thereof] in the clinician.
Because this article is, in essence, just another opinion piece, let us then focus on logical reasoning and objective arguments. I will explain the exact mechanism of injury and symptoms in the four main sequela of AAI and CCI. Hopefully, this piece will prevail in explaining logical arguments for legitimate findings in CCI and AAI, and therein lead to a gradual decline and prevention for related misdiagnosis. Ultimately, the reader must discern for themselves.
As mentioned initially in this article, craniocervical instability is mainly associated with jugular outlet obstruction and basilar invagination, whereas atlantoaxial instability can cause posteriorization of the dens and brainstem compression, or rotational dysfunction resulting in either bow hunter’s syndrome, Cock Robin syndrome or other variants of segmental luxations. Let us look closer at these clinical entities and their associated symptoms, imaging findings, and, importantly, clinical triggers.
For the sake of relevance, this article will mainly address ligamentous and muscular injuries, as these topics, especially when mild, are much more controversial than incidences of CVJ fracture. Information about the identification of CVJ fractures will not be applicable for patients with chronic workups and lacking imaging findings over a long period of time.
AAI/CCI – Joint [sub]luxations
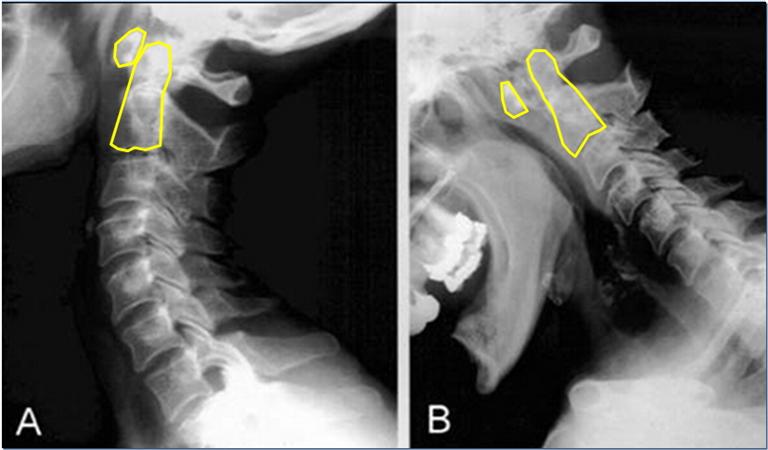
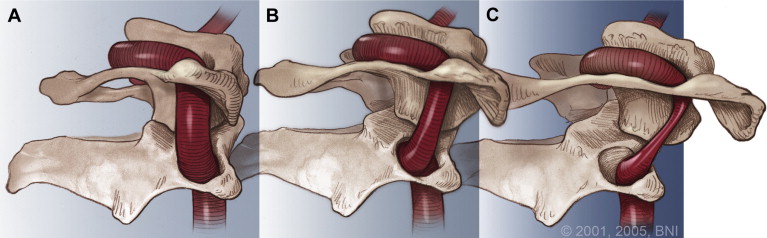
In circumstances of gross trauma, the ligamentous damage may be so severe that the entire vertebrae luxate (dislocate) from normal position. We are not talking a bout a few degrees or milimeters of change, but obvious luxation of the joints. This is easily seen on imaging, especially on CT, as the alignment of the joint will be unequivocally abnormal to the extent that would not be achievable without tremendous ligamentous injury. The atlas can sublux anteriorly, posteriorly, laterally, or vertically. More commonly, however, a due to asymmetrical tearing of the covering ligaments, rotational subluxation or frank luxation is seen according to the Fielding & Hawking classifications (1977): Type 1, 2, 3 and 4, wherein types one and two are the most commonly encountered ones. Type one involves sole rotary luxation of the facet joints, usually along with damage to either the alar ligaments and capsular ligaments. Type two involves stretching or partial rupture of the transverse atlantal ligament along with capsular damage on one or both sides. Type three involves anterior subluxation of the entire atlas due to combined full rupture of the TAL and partial rupture of the capsules and other structures. Type D would generally involve a dens fracture as the atlas migrates posteriorly, along with facetal luxation and capsular rupture. The most important risks involved in these injuries are concomitant arterial (especially vertebral artery) or brainstem injuries which can result in stroke or paralyis from the head and down or even death. Patients with rotary subluxation will develop torticollis and will generally appear fixed/rigid upon physical exam and may not be able to rotate their necks at all. Surgical reduction and fixation would be the only appropriate treatment.


Vertical and horizontal [sub]luxations
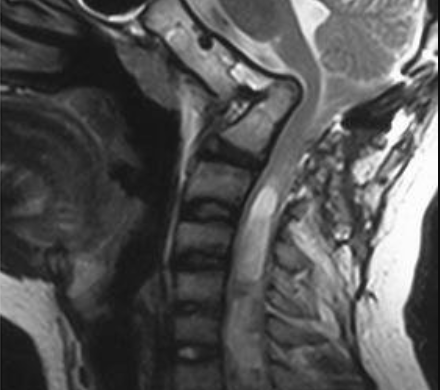
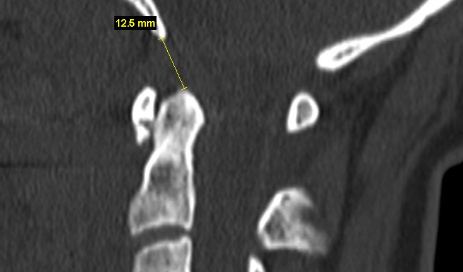
Aggressive craniovertebral junction ligamentous injuries can also result in vertical displacements. Typically, complete membraneous ruptures of the CVJ may cause dislocation between the head and neck, resulting in positional dissociation between the the two. This will be predominantly evident on a flexion/extension scan, where the basion-dens interval (BDI) will be dynamically increased, and greater than 10-12mm (Ross & Moore, 2015; Deliganis et al. 2000). Complete rupture of the transverse atlantal ligament, however, will generally promote dorsal and cranial migration of the odontoid process (the atlantodental interval (ADI) will be increased (> 3,5mm) while in flexion) causing it to compress the brainstem dorsally (in the upper neck), or to migrate into the foramen magnum and compress the brainstem there (basilar invagination), where the tip of the odontoid will be seen far above the Chamberlain’s line, whereas it in normal patients sits about 2mm below the line. This can also promote anterior dissociation of the head which will lead to an abnormally high basion-axial interval (BAI – Harris measurement) of more than 12mm (Ross & Moore, 2015). In some circumstances, gradual degenerative basilar invagination can also occur due to gradual and progressive degenerative horizontal misalignment of the atlantoaxial joints (Goel 2014), due to certain diseases such as rheumatoid arthritis, but it is usually caused by head and neck trauma. Powers ratio will be abnormal in cases of both BI and craniocervical dissociation (Ross & Moore, 2015). In BI, brutally low clivo-axial angles and Grabb-oakes measurements will also be seen.

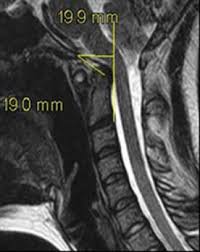
In vertical dissociation of the CVJ, the main dangers will – similarly as above – involve potentially dangerous pulling and pushing on the blood supply to the brain (carotid and vertebral arteries) as well as the brainstem itself, potentially causing dissection of the arteries. The abnormal imaging findings will mainly be evident during extension of the head and neck. Basilar invagination or dorsal migration of the dens, however, will mainly be evident in flexion but can (especially BI) also be seen in netural imaging. These problems will mainly endanger the brainstem. If combined with Chiari malformation, compression of the cerebellar tonsils can cooccur and will occur with lower measurements than normally needed to cause brainstem compression alone, due to filling of the space behind it (the descended cerebellum).
Lateral [sub]luxations
Lateral bowing of the inferior atlantal facets in netural position is a sign of transverse atlantal ligament laxity. It will rarely cause frank luxation, however where the facets dislocate and lock laterally. Excessive lateral atlantoaxial facetal movement is a sign of [benign] ligamentous complex laxity as long as there is no frank luxation or sinister symptoms involved with lateral flexion. The ligaments involved are the transverse, alar and capsular ligaments. Burry et al (1978) documented a rare case of lateral luxation in a patient with rheumatoid arthritis, in which the supporting facet had eroded away. Musa et al. (2019) documented another case where a patient with RA developed odontoid fracture and subsequent anterolateral subluxation of the atlantoaxial joint. Generally, however, in ligamentous laxity, some bowing and lateral hypermobility (evident by lateral flexion “overhangs”) will almost definitely not result in frank luxations down the line nor do they tend to elicit symptoms from the actual atlantoaxial facet joints.
Imaging vs. clinical
It is imperative to understand that patients with dagerous craniovertebral junction injuries, although one may sometimes require a dynamic CT or x-ray to identify them, will have clear imaging findings combined with clear clinical triggers in the utmost majority of incidences.
A patient with positional brainstem compression due to TAL rupture, for example, will develop neurological (ie. medullary) symptoms when looking down, and will tend to improve when pulling the head up and back. If the brainstem compression is not positional, ie., it is seen even on neutral imaging, then the symptoms would be expected to be constant. Symptoms of brainstem compression are respiratory crisis and quadriplegia, but can also manifest more diffusely. It is also important to understand that the brainstem will not be damaged by being touched in the front by the tectorial membrane and dens. Rather, it must be compressed by the dens ventrally, and flaval ligament and lamina posteriorly. I have seen several patients misdiagnosed and become almost paralyzed by anxiety due to an increased Grabb-Oakes measurement where the dens is just barely in tangent with the brainstem, despite zero evidence of actual compression nor signal changes in the brainstem — and with normal neurological examinations without any upper motor lesion signs! This iatrogenic practice must come to an end.
Patients with severe ligamentous compromise and a risk for actual dangerous secondary potentially pathologies, must have instability so aggressive that it can cause damage to the brainstem or adjacent cerebro-arterial supply. Facetal locking with rigid torticollis (Cock Robin syndrome) or similar, in cases where there is no neurological compromise, is less dangerous. Albeit still a surgically treated problem.
Thus, the patients in the rotary subluxation group are expected to present with severe and sudden neck pain as well as rigidity to the extent of being unable to move the neck. This, with or without accompanied neurological symptoms, be it vascular or neurological. A 3D rendered CT scan should easily demonstrate the luxation in cases where the sagittal slices appear normal or close to normal, whereas cases of dens migration will also appear obviously abnormal in the sagittal planes of imaging. Sometimes flexion-extension and rotational imaging is necessary.
Patients with horizontal instability of the craniovertebral junction but without rotary subluxation may not necessarily demonstrate the same level of rigidity, but may show induction or resolution of symptoms as they venture into flexion vs. extension. This would depend on whether or not the compression of the brainstem is constant, which again would depend on several factors. In BI, the compression tends to be constant. The patient should demonstrate some brainstem symptoms, and may develop quadriparesis if the compression is sufficiently hard and constant. Imaging will prove brainstem compression on [flexion/extension] MRI, and an increased atlantodental interval on flexion/extension CT or x-ray. Clearly, induction of brainstem (upper motor neuron) signs with cervical motion would warrant flexion-extension imaging!

Risk in asymptomatic patients: If the patient has craniovertebral dissociation either due to anterior or superior migration of the head in relation to the cervical column, one may argue that there is a risk for traction injury to the brain’s blood supply even in cases where the patient has no obvious induction of symptoms upon flexion-, extension or rotation, and has no imaging that demonstrates neurovascular conflict (eg., BHS or positional brainstem compression). In such a case, however, certain important measurements (not mere CXA (norm: 150-180 degrees) or Grabb-Oakes (norm. <9mm), which overestimate the pathologies and are much misunderstood due to unrealistic consensus of what is “normal”) will clearly be abnormal, such as the Harris measurement (BAI), basion dens interval (BDI), or Powers ratio. If the measurements are within normal limits, the likelihood of dangerous sequelae are low, if not absent. This would apply for patients with obvious hypermobility but who do not have clinical triggers compatible with CCI or AAI (induction of symptoms in flexion, extension or rotation, and complete normalization when in neutral). For patients with post-traumatic ligamentous injuries where measurements are still within normal limits, obvious segmental effusion should be seen despite otherwise normal anatomical positioning. Dissection of the vertebral and carotid arteries is fairly rare and can be excluded through a doppler ultrasound or CT angiogram.
AAI – Bow hunter’s syndrome
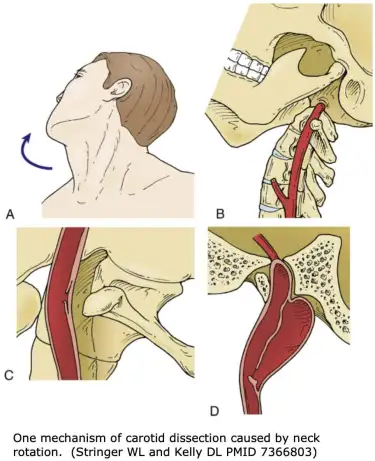
Patients with hyperrotation of the atlantoaxial joints can also develop Bow hunter’s syndrome (BHS). BHS implies rotational compression of the vertebral arteries, which are two out of four arteries that supply the brain (two internal carotid and two vertebral arteries). This can happen due to excessive rotation at the joint with gradual worsening (eg., in a patient with Ehler Danlos syndrome or similar), or in combination with rotation and transverse-foraminal stenosis, which is the hole on the side of the transverse processes that the vertebral arteries and veins venture through. Some rare cases have also demonstrated rotary compression of the vertebral artery in the lower neck due to arthritis or disc bulges that fills up the transverse foraminae (Ujifuku et al. 2009), but this is extremely rare.
One is especially predisposed to this problem if the affected vertebral artery is highly dominant (much higher caliber than its contralateral counterpart) or if the contralateral artery is extremely hypoplastic, or, finally, the contralateral artery terminates as the posterior inferior cerebellar artery rather than at the basilar artery (Josy & Daily, 2015). Most cases of mild to moderate – unilateral – compression, sometimes even intermittent occlusion, is asymptomatic due to contribution from the contralateral VA (Faris et al. 1963). If the patient is indeed positionally symptomatic, however, and there is compatible imaging evidence, either atlantoaxial fusion, transverse foraminotomy or certain physical therapies may be warranted depending on how severe the findings and symptoms are.
Patients with genuine and symptomatic rotational vertebral artery compression will develop symptoms of vertebrobasilar insufficiency when they fully rotate their heads to one or both directions, and may be further worsened if done simultaneous with neck extension (DeKleyn 1927). The symptoms will completely resolve when returning to neutral position; usually even a few degrees reduction is enough to normalize flow. This is important to understand, because maximal rotation will induce, and neutral position will stop the symptoms in patients with legitimate vascular conflict in AAI. Symptoms of VBI develop rapidly in patients with legitimate and adequate degrees of vertebral artery compression when placed in the triggering position. In addition to reproducible clinical triggers (positions), the patient should preferably undergo a dynamic catheter angiography of the neck. If unavailable, a CT angiogram can be used, but is less sensitive. I don’t recommend MRA. The findings may be quite subtle and are easy to miss outside of dynamic exams.

CCI – Jugular outlet obstruction
Jugular outlet obstruction is commonly seen in patients with upper cervical horizontal facetal misalignment, and especially if they have broad transverses processes or a posteriorly angulated styloid process (Gweon et a. 2011, Dashti et al. 2012). This is really more of a poor posture/misalignment problem than a case of instability (Larsen 2018), but because it is a legitimate upper cervical problem then I will still mention it in this article. Just anterior to the transverse process in patients with normal necks, emerge the internal jugular veins as well as the glossopharyngeal, vagus and accessory nerves. Horizontal misalignment of the facet joints often cause dorsal migration of the C0 and C1 facets which cause approximation of the styloid process and the C1 transverse processes. This, seriously augmented by poor “hinge neck” postures (Larsen 2018). Both positional (ie., upright. Early stage) and constant compression (if seen on mri, moderate, if seen on CT, severe) of these structures may occur.
The renowned scholar and neurosurgeon professor Atul Goel was the first person, to the best of my knowledge, to acknowledge and document the notion of horizontal misalignment of the craniocervical facet joints and that this would often be present despite a completely normal-looking mid-sagittal slice (where most craniovertebral junction measurements are done). He also found that severe misalignment of these joints were often associated with Chiari malformation, basilar invagination, and various other pathologies. Seemingly unrelated, Higgins et al (2013) and others (Dashti et al 2012, Li et al. 2019) have documented numerous symptomatic cases of jugular vein stenosis at the craniovertebral junction. To the best of my knowledge, I was the first person to document the notion that this was, in essence, a postural phenomenon that is induced due to poor posture over a long period of time (Larsen 2018). The problem has received various names such as mere “jugular vein compression”, “venous eagle’s syndrome”, but I have called it “jugular outlet syndrome” (JOS), as it is a problem that not only affects the craniovenous outflow, but also several cranial nerves, and can be culpable in various strange neurological disorders (Read my atlas article (link) I also have an upcoming paper on this topic that I hope to release this or next year).

Patients with craniovenous outlet obstruction due to JOS may induce their symptoms with a Queckenstedt’s test, that is in essence a manual compression test of the internal jugular veins. A positive test would be interpreted by unbearable head pressure, lightheadedness, worsening of headache, etc., within about 20-30 seconds. Followup, as mentioned above, can be a CTV, volume flow doppler exam, and potentially catheter venography and manometry as one additional confirming pre-surgical step to ascertain actual raised intravenous pressures. In my experience, we would expect to see at least 20mmHg maximum venous pressures. Presuming the central venous pressure being normal, then I am not so interested in the pre and post-stenotic gradients as they tend to be unreliable. I prefer to compare mid-jugular to the highest pressure found, usually in the torcula or SSS.
Compression of the glossopharyngeal nerve will frequently cause pharyngeal pain (back of the throat pain) whereas vagal compression may lead to dry coughing, “lump” in the throat feeling, ear itching and various strange things when unilateral, but has been associated with more problematic issues when bilateral such as gastroparesis (Waldock et al. 2020). Accessory nerve compression can cause weakness of the trapezius and sternocleidomastoid muscles, but can also cause cervical dystonia. I have also seen cases of seventh nerve dystonic mimicks several times in JOS, where platysmal dystonia or even oropharyngeal dystonia (hypoglossal nerve) has been identified, worsened with neck tucking (which increases the compression) and resolved with specific strategies for widening the atlanto-styloidal interval (see my atlas article as linked earlier) or Larsen 2018 in the reference list). The same applies for conservative strategies to reduce internal jugular vein compression.
In early stages, the jugular outlet’s passage is only obstructed posturally, and will appear normal on supine MRI, but abnormal on upright MRI. In moderate stages, the MRI will appear abnormal, but the CTV will still appear relatively OK (because the patient tends to be placed on a neck wedge which protracts the head in the CT machine — this reduces the compression). In late stages, even the CTV will show severe compression, and at this stage, surgery may be the best option for resolution if there is clinical correlation. Mild to moderate cases tend to respond well to appropriate conservative therapy (not general therapy), cf., once again, my atlas joint article from 2017 linked several times earlier.
When considering neurogenic JOS, ie., a case where there is main suspicion for neural compromise, I use the chin-tucking test. The patient will hinge back at their neck while simultaneously flexing the cranium. The deep neck flexors should not engage as this lessens the compression. Stay put for 30-60 seconds, look for worsening of symptoms while in the test. Sometimes, the symptoms may trigger within a few minutes after the test as well, depending on various factors which exceed the scope of this article. Either way, if positive, move on to confirm narrowing of the jugular passage between the styloid process and C1 transverse process on a CT scan. A CTV is preferable, but a general neck CT will also do if you have sensitive kidneys and would like to avoid contrast infusion.
As stated, although rooted in postural dysfunction, this is not really a problem of pathological instability, and therefore I don’t recommend neck fusion to treat this problem. In many circumstances, conservative treatment (Larsen 2018, atlas joint article as linked earlier) is appropriate. In severe cases, I recommend postural corrections (appropriate, not generic) along with styloidectomy and transversectomy. It is, technically, possible to perform traction, reduction and fusion to obtain the same result, but this would be like killing a fly with a canon. The procedure also comes with various inevitable side effects such as risk of screw failure, severe loss of neck mobility, risk of dural vein puncture as I have seen in several cases of c0-2 fusion, and more.
Illegitimate and non-sinister incidences AAI / CCI
As touched upon in the beginning of this article, that prompted me to write this article, is a huge – massive – influx of patients over the last few years who have been illegitimately diagnosed with AAI or CCI. The utmost majority of these patients have have normal supine imaging, and many of them also normal or nearly normal upright imaging. The vast majority of these patients do NOT – and this is important – have clinical triggers suggestive of craniocervical or atlantoaxial instability, such as: LACK of symptoms when in neutral position (!), induction of symptoms (all or nearly all of your symptoms, not “some neck pain”) with maximal rotation, nor during flexion or extension. Flexion and extension imaging fails to demonstrate any sort of brainstem compression.
Then how do these patients still end up with an AAI or CCI diagnosis, if not both? First, need I mention the notion that there is tremendous money in this patient group, and that if treatment goes wrong, becuase they have already burned their bridges with their GPs, no one will listen nor care? This may not apply for all of them, but it is a common problem which makes this patient group especially susceptible to become perfect victims of medical vulturism. Second, because it is such a controversial topic that lacks medical consensus, poor understanding of the actual mechanism of pathology leads to misunderstandings. For example, although the medical literature (almost exclusively biased reports written by people considered experts on the topics (I am also biased on the topic; all experts are) may suggest a clivo-axial angle lower than 150 degrees as abnormal, this is still a measurement used to associate concrete craniocervical angles with medullary compression. If there is no medullary compression, not even in a flexion/extension scan, then we cannot say that the patient is of surgical degree, even if it is very low, unless they look so bad that it is reasonable to expect frank compression in the near future!
It baffles me when I see patients with 130 degree CXA and some additional signs of mild/moderate laxities being butchered with C0-T1 surgery despite there being NO instability in the cervical spine and only mild findings in the upper neck that are not causing any neurovascular conflicts nor facetal lockups (eg., Cock Robin syndrome). This is not good medical practice. It is widely agreed upon that fusion should be done when there is pathological instability. What does this mean? It means that the instability is, or will probably, shortly, become bad enough to carry the potential to damage nerves or blood vessels. For example, if there is a C4-5 anterolisthesis with resultant chronic radiculopathy, C4-5 ADCF would often be utilized as operative treatment. If there is a 1mm listhesis, however and the patient has no neurological symptoms and the medulla is utterly free of compression, then performing fusion is completely unnecessary. The same principles would apply for AAI and CCI: There must be clear imaging findings, and I am not talking about a simple measurement being off, but real pathology proven to be associated with the given diagnosis. If the patient turns their head and passes out, and a catheter scan demonstrates dominant vertebral arterial compression, then certainly this is a case of AAI and atlantoaxial fixation may be a viable option, at least if the transverse foraminae are normal. But a patient who just “feels bad” (even if they feel very bad), and especially if they do not have positional triggers and their imaging does also not demonstrate constant brainstem or otherwise vascular compromise that fits with the symptoms, then diagnosing such a patient with CCI or AAI and claiming its presence as the culprit of their symptoms, is madness. Please understand that no matter how bad you feel, pursuing the wrong diagnosis will not help.
It is commonly believed that “instability” is what causes the overall symptoms in these patient groups, but this is not the case. The reason why AAI and CCI are – potentially associated – with so many symptoms such as headache, dizziness, etc., is due to the potential for neurovascular conflict. It is not due to mild overall instability that does not cause neurovascular conflicts.
Clunking, clicking and pain in the upper neck
Clunking and popping that occurs in the upper neck can be scary, but is usually just a sign of facetal rigidity with reduction, meaning that they get stuck and then pop back into place. Facetal rigidity and dysarticulation is very common in patients with poor cervical postures and functionality of the neck muscles, and especially the muscles that restrict rotation and attach directly onto the spinous or transverses processes in the spine. When these muscles get tight (due to profound weakness), due to poor posture and movement patterns, or, as well, in many cases due to head or neck trauma, restricted joint movement will occur and popping and cracking, even loud clunks can occur. However, as stated, in most cases this is just locked facets that suddenly reduce (realign) with a pop.
If the symptoms happen along with aggressive neurological symptoms, however, or if your neck locks up in rotary fixation, greater concern could be applicable. But this is rarely the case in my experience. If it is, however then flexion/extension and rotational imaging to exclude positional facetal luxation is warranted. Dynamic angiograms could also be applicable in certain circumstances, cf. the section on bow hunter’s syndrome. You can also get these images done to get peace of mind if you do not have strong neurological sequelae related to the popping, but beware that many of these “specialist clinics” diagnose AAI CCI no matter what your imaging looks like, and therefore I generally recommend working with larger hospitals. A general neck MRI is usually a good idea and may show some arthritis in the atlantoaxial and atlanto-occipital joints along with minor intra-articular effusions, suggesting irritation of the joints. This is not dangerous, but can cause some popping, restriction in movement, and some pain upon articulation.
For treatment of the facetal dysfunction I recommend postural correction for the head neck and shoulder blades, along with exercises for the trapezius, levator scapulae, suboccipital and deep neck flexor muscles. See my youtube channel for appropriate training. Training is done carefully twice per week. Posture is done for the rest of your life. See my other articles or YouTube videos for howtos.
A lof patients have clicking and clunking in the neck along with severe suboccipital pain. Beware that suboccipital pain, espeically if your imaging is normal, is a very common sympton in thoracic outlet syndrome, and is actually a migraine variant. This pain tends to get worse with stress and with high heart rates, and are often also worse in the morning after lying down. This is a component of TOS CVH in most circumstances, in my experience, but can certainly scare the patient into believing that they have sinister CCI or AAI due to the location of the pain along with heavy cracking and other symptoms.
Although the complete differentiation between this and CCI or even occipital neuralgia is something that is complicated and must be done on individual basis after examination, we can, in essence, say that suboccipital pain that worsen with shoulder loading tends to be TOS or occipital neuralgia, whereas suboccipital symptoms that induce when lying down or being upright regardless of neck position tends to be TOS CVH. Suboccipital symptoms that occur only with cracking, if the MRI shows arthritis or joint effusion, especially if the neck locks in rotary fixation, then this could be a case of legitimate AAI or CCI. For occipial neuralgia, an ultrasound guided nerve block will cure these symptoms for three hours and thus confirm the diagnosis. For TOS CVH the patient will generally feel better when stress is reduced along with taking beta blockers (confer with your doctor). Patients with AAI CCI will be expected to trigger symptoms only with neck movement (being upright alone is not enough) and resolve (fully) when the neck is held still.
Commonly misunderstood and overemphasized measurements
I hope that, by now, the reader has understood the importance that clinical measurements, actual pathology and clinical triggers should go hand in hand. If the patient has an elevated Grabb-oakes interval of 10mm and low CXA of 130 degrees, there is some horizontalization (upwards deflection) of the medulla, but no compression from both sides. It is, as we say, “in tangent” with the dens and tectoral ventrally alone. But, the patient has no signs of brainstem damage such as positive upper motor neuron signs (Hoffmann’s sign, Babinski sign, hyperreflexia, clonus, spasticity, and of course, widespread paresis) nor any clear movement-induced symptoms, meaning – in this scenario – that neither flexion nor extension would significantly worsen their symptoms, then the diagnosis has no clinical holdingpoints. Now, what if there is no frank compression nor clinically medullary signs and triggers, but there is a very small space both infront and behind the medulla that has been gradually getting worse. In such a case, to avoid foreseeable medullary damage, one may reasonably opt for fusion as preventative surgery, because the medulla, once damaged, does not always recovery after surgery. This is reasonable. But if there is lots of space for the medulla, such invasive surgery simply is not warranted.
Another scenario could be that the patient has been diagnosed with atlantoaxial rotary subluxations, as little facetal overlap, let’s say, 15%, is seen upon bidirectional rotation. Two important questions arise: Does the patient actually develop (even if just from time to time) develop frank facetal luxations causing the neck to lock up? If not, does the patient actually have any significant symptom induction with rotation? And if yes, do they completely normalize when resuming neutral position? Remember that the main dangers of atlantoaxial hypermobility are 1. facetal luxation, and 2., risk for rotational injury to the vertebral artery. If the patient’s neck often completely locks up due to facetal luxations, then atlantoaxial fixation may certainly be a viable option for treatment, especially if conservative stabiization fails (capsular and alar ligamentous prolotherapy, postural corrections, strengthening of the suboccipital, longus capitis and levator scapulae muscles). However, if the patient has symptoms regardless of being in rotation or not, and has never had a case of alantoaxial rotary fixation, then there is no evidence that this is the cause of the patient’s symptoms, even if it, indeed, may be a bit loose. In such cases I tell my patients that, yes, you do have mild AAI, but it is not causing your symptoms. We can still treat it preventatively, but it won’t resolve the symptoms.
Another problem with regards to rotation, is that the measurements are often done wrong. I very often receive upright MRI reports where the rotation is completely normal, and the patient is still diagnosed with AAI. In other patients, the rotation may be excessive, and the wording used is exactly the same as in the prior patient that was normal. What I prefer to do is to first draw lines that show the actual rotational alignment of the C2 and C1 when looking left and right. It is important to understand that the size of the facets is what determines what degree of rotation would be excessive. For example, I have seen patients with 45 degrees of rotation (which is higher than normal) between the C1-2 that had completely normal overlap due to large facets, and I have seen patients with 30 degrees of rotation (which is usually completely normal) with poor overlap and AAI, due to small facetal surfaces. Thus, it is important to measure both the percentile overlap as well as the degree of rotation bidirectionally. In most circumstances, even if there is poor overlap but no evidence of frank facetal luxations (clinical history or with provocation), then conservative therapy can usually prevail in management.
I have lost the count of the amount of patients, terrified patients, who have been scared by diagnoses such as brainstem compression with zero evidence to support it. One patient was told by a famous alternative european neurosurgeon that she has CCI and AAI, and although there is no evidence for current surgery, she would probably be in a wheelchair within a few years and might even die. … Although there were no current grounds for surgery? Then, if there are not even sufficient findings for surgery, how can one possibly give such a fatal prognosis? Conveniently, she was sent out to a colleague for very expensive nonsense therapy (again, regardless of lacking serious findings that would require surgery) and sent tens of thousands of euros on stemcell and prolotherapy procedures in a desperate attempt to avoid the inevitable wheelchair. Another patient was told by a well-known pain physician in the US that she had brainstem compression and required several expensive prolotherapy procedures. When I reviewed both of these patients’ imaging and cases, the only findings were slightly low CXAs and a Grabb-Oakes around 9mm. Both patients had severe symptoms regardless of lying down, wearing a neck brace, etc., and did not get worse nor better when turning or moving their necks. The brainstems were completely void of evidence for compression in both cases, and there was no evidence of signal changes (consistent with brainstem damage) on MRI. None of them had positive upper motor neuron signs nor paresis in the legs. Now, the “I was told” is clearly second-hand information, and I cannot guarantee its accuracy. Regardless, both patients were terrified and thought they would end up in a wheelchair, so it sounds quite believable to me. Moreover, I have heard numerous similar stories from other patients. This madness must stop. At the very least, if the clinician has clinical suspicion but no concrete holdingpoints for their diagnosis, they must be honest about this. Then the patient can make an informed decision about whether or not they want to invest in experimental therapy.
Thus, beware that a low clivo-axial angle (CXA) is often overinterpreted and abused as supportive evidence. Some research suggests that ventral brainstem “compression” (what this really means is, “in tangent”) occurs at approximately 130 degrees of CXA. Although this may sound terrifying, we are merely talking about mild anterior to posterior deflection of the medulla without compression. To compress the brainstem it must be compressed from both sides, both infront and behind. Medullopathy (signal changes, cord damage) will not occur by mere deflection, which is also evident by the blatant lack of upper motor neuron findings in these alleged “brainstem compression” patients. I have seen patients with a CXA as low as 110 degrees and still did no have any frank brainstem compression. Therefore, when I hear about patients being operated on with no other abnormality than a CXA of 140 degrees, my opinion is that this is reckless butchery. Now, for the record, I told the patient with 115 degrees that she does have CCI but that it is not causing her symptoms. However, I also told her that she may end up having fixation surgery in the future to prevent foreseeable compressive damage to the brainstem. This, of course, must be evaluated on a case-to-case basis. Another common belief is that this mild deflection stretches the brainstem and somehow causes damage. If this was the case, ie., if the brainstem and medulla was being stretched, then the patient would highly likely get neurological symptoms that improve with extension and worsen with flexion (as patients with legitimate tethered cord syndrome do), and would certainly have a positive Slump test, a test which stretches the spinal cord. This, however, is very rarely the case with this patient group in my experience. Furthermore, a claim of brainstem stretching and kinking with resultant medullary microdamage that somehow not responds negatively to being stretched in real-time, and also lacking upper motor neuron signs, is not a very realistic claim.

Grabb-Oakes interval is another measurement that is often misunderstood. Just like the CXA, this measurement is supposed to aid with objective measurements rather than just “eyeballing” the images, and writing down your impressions. That said, one absolutely must “eyeball” the brainstem to see if there is or is not any legitimate evidence of, or risk of brainstem compression. For example, if the brainstem is compressed due to a ruptured transverse atantal ligament or due to basilar invagination, a brutally high Grabb-oakes measurement would be expected, and would be a nice extra detail in the report along with the actual information that there is indeed anterior-posterior compression of the brainstem. But this measurement in and by itself, when it is 9 or 10 or even higher, but there is no brainstem compression – not even in flexion-extension imaging – this cannot be interpreted as a surgical indicator. Rather, just like with the CXA, it is an indication of the present spinal health status and perhaps also an indicator as to non-surgical prognosis as well as an indicator of likely outcome if nothing is done. If the patient has a Grabb-Oakes of 18mm, however, and the transverse ligament is ruptured with the dens compressing the brainstem from the front and pushing it into the lamina behind it, then this is an emergency that requires timely surgical decompression. In such a case, UMN symptoms and signs would be expected as well.

BDI, ie. the basion-dens interval, is the distance between the tip of the clivus and tip of the C2. In other words, the vertical distance between the head and the spine. Research has shown that normal limits are 3 and 10mm, with an absolutely maximum of 12mm (Ross & Moore 2015). Something I often see reported as alleged evidence of sinister CCI, is a translational BDI or BAI (the basion-axial interval is the horizontal distance between the tip of the clivus and the posterior wall of the odontoid process. The BDI indicates vertical-, and the BAI horizontal structural integrity. Both measurements tend to worsen with neck extension. The surgeon may claim that because there is translational differences, meaning that the interval increases with movement, this is evidence of sinister CCI or AAI regardless of the measurement still being within normal limits. I completely disagree with this and, once again, refer to common sense thinking that if the joint positions are within normal limits then there is very little risk, if any, of any damage to the spinal cord or segmental arteries. This, once again emphasized if the patient also does not induce any sinister symptoms in the positions where the alleged instability occurs. It does certainly insinuate some instability and ligamentous laxity, and can certainly result in greater level of wearing and tearing of the facet joints and causing some neck pain and joint effusions, but it can not be said to be any form of sinister AAI or CCI due to lacking neurovascular conflicts. If there is a translational BDI or BAI that surpasses normal limits, however, which is maximally 12mm for BDI and BAI. A caveat here may be if the the translational value is very high, as this would be a reasonable indication of foreseeable joint damage, but there is no consensus in the literature with regards to how much that is. Some have proposed 2mm of translational difference, but this is completely unreliable in my opinion and exprience. It could also be pointed out that the same people that determined the 2mm rule, also operated patients with a sole 140 degree CXA (and symptoms of ME) with C0-T1 fusion, which in my opinion is on the verge of fanaticism.
Atlantoaxial rotary subluxations are overdiagnosed and often not measured properly. I recommend first measuring the degree of rotation between the C1 and C2 by drawing a line from the bifid process to the middle of the anterior aspect of the vertebra, and then another line from the posterior to the anterior tubercles of the C1. Compare the two to obtain the degree of rotation. Secondly, and perhaps more importantly, the extent of facetal overap must be measured. When rotated to the right, making sure that the axial alignment of the imaging program is aligned with the spinal column longitudinally, compare the anterior aspect of the right facet vs. the facet of the C2, and the posterior aspect of the left facet vs. the facet of the C2 and calculate the actual percentile of overlap. I see massive amounts of patients with alleged AAI who have normal atlantoaxial facetal overlap, and of course, also lacking clinical correlation.


Signs of ligamentous damage. This is one of the biggest offenders along with DMX and CXA, causing massive confusion, coercion, and misdiagnosis. Patients with normal structural alignment and more or less normal or completely normal radiological imaging, without clinical correlation, end up diagnosed with CCI or AAI due to a slightly low (non-sinister) CXA, say 135 degrees, and some signal changes in the alar ligaments on T2 FLAIR imaging or slight increase in the atlantodental interval (ADI) despite normal thickness of the transverse atlantal ligament (TAL). First of all, studies have shown that FLAIR hyperintensities (suggestive of ligamentous partial rupture or damage) have been found in a lot of asymptomatic patients (Myran et al. 2008). Second of all, if there is suggested ADI widening, but a high quality supine MRI with low slice thickness ascertains patency of the majority of the fibers of the TAL, the likelihood of actual complete rupture and future brainstem injury is extremely low. Therefore, when there is evidence of equivocal findings such as signal changes in ligamentous structures without expected adherent findings such as gross hypermobility compatible with the injury at hand, this can generally not account as someting sinister. However, if there is obvious compromise of a ligament but there is no evidence of sinister hypermobility or structural displacement (eg., very high ADI), the ligamentous should be further examined with high-resolution T2 FLAIR imaging with low slice thickness (supine imaging!) to get a better impression of its actual thickness. Upright MRI has very low quality and because of this, there is a lot of guesswork involved in its interpretation. Followup with a dynamic CT, supine MRI or similar to confirm potentially equivocal findings is warranted.

DMX. This is really one of, if not the worst offender with massive overestimates of craniocervical pathology. I have seen countless reports from DMX centers where the patient, despite having normal or virtually normal conventional imaging, the patient is delivered reports of laughable quality, typically deeming the whole neck as unstable, despite the images being virtually normal. Claims of three, four or even five-level spondylolisthesis due to a 50 micrometer (0.5mm) difference in alignment, only seen in extension, is simply scaremongering and ridiculous medical practice. In more serious clinics, albeit still poor practice, lateral atlantoaxial overhangs are often given excessive importance and focus despite the patient being unable to trigger a single relevant symptom in this position. Due to the poor practice integrity that is often associated with DMX imaging, despite these modalities indeed having some utility in certain cases, I cannot recommend having them done unless done in a serious hospital without a financial incentive (ie., without financial connections to the clinician ordering them), and without a very obvious scope of investigation that could not already be seen in MR or CT imaging.


Required imaging
If you are very concerned that you have craniocervical and atlantoaxial instability, then I recommend getting workups for both these — but also relevant differential diagnoses.
For the AAI / CCI:
The main scope of the below studies is to 1. exclude neurovascular conflict, and 2., to look for legitimate signs of instability be it with or without neurovascular conflicts, in order to determine degree of affliction, prognosis, and treatment plan. And, of course, to determine whether or not the findings actually correlate with the patient’s symptoms and clinical exam. Finally, beware that many of these uMRI clinics render horrible images that barely show any anatomy, yet somehow still manage to determine various complicated diagnoses from them. I recommend sticking to clinics that have good reputations and good imaging protocols.
- Supine cervical MRI including T2-w sagittal-oblique sequences at 2mm slice thickness (disc and foraminal health is best evaluated on a supine MRI).
- Upright cervical MRI in flexion, extension and maximal bi-directional rotation. In my experience, although I usually disagree with their diagnoses, is that Medserena in London has the absolute best upright imaging quality in the world. I have not receiving anything that comes close of what they produce. (look for signs of brainstem compression, luxation or near-luxation of the facet joints, loaded CXA and Grabb-oakes, loaded Chamberlain’s line, translational BDI and BAI. Look for upright compression of the IJVs)
- Dynamic CT also works well, but has much more radiation. Dr. Gilete in Spain, although I often disagree with his diagnoses, tends to order beautiful dynamic CT scans and also good craniovascular scans. (look for the same things, as well as loaded and positional narrowing of the atlanto-styloidal spaces, the latter only being visible on CT)
- DMX — I don’t recommend getting a DMX. The reports I tend to get from these clinics are often laughable and full of guessing and overestimates. I am not saying that this applies to every DMX center nor that DMX in and by itself is never useful, but due to the overwhelming lack of competence that tends to come with these studies, I don’t recommend them unless unless you have obviously abnormal imaging otherwise and want to look for occult fractures or similar sinister and stubbornly identified problem.
My experience is that most of these patients suffer from craniovascular pathologies, not CCI and AAI. That is why they are much less affected by actual neck position than legitimate CCI AAI patients are, and certainly do not become symptom free in neutral positions. Thus, I recommend the following studies for craniovenous hypertension and TOS CVH:
- Head MRI (look for signs of elevated head pressure, beit vascular or CSF related. Common findings: Ovalization of the orbitae, dilated optic nerve sheaths, pituitary concavity, Chiari malformation, “tight brain” appearance, jugular vein compression with or without white-vessel signs, dilation or narrowing of the lateral and possibly third ventricles, periventricular ependymal T2 FLAIR hyperintensities)
- Neck MRI (general evaluation of the neck integrity)
- CT angiogram of the head neck and subclavian arteries with the arms raised (contrast infusion via femoral vein. If nicely timed, around 20 secs after infusion, beautiful visualization of both arteries and veins is permitted). Look for jugular vein compression, dural sinus and neck vein integrity, exclude typical patholgies such as aneurysms etc., exclude vertebral or carotid dissections, evaluate the thoracic outlet for interscalene, costoclavicular or subpectoral stenosis)
- Doppler of the carotid and vertebral arteries (look for signs of hypertension, cf. our TOS CVH paper (Larsen et al 2020). This is a major component in the workup for TOS CVH)
- Fundus exam (must be properly zoomed, must be exported in high digital quality and resolution). Look for signs of retinal hypertension (subtle copper wiring, AV nicking, tortuosity of the arterioles, generalized vasospasm or papilledema. I recommend doing this with a neuro-ophthalmologist, not a general ophthalmologist or opticician, as the findings are often missed.
Craniovasculo-hypertensive disorders (mainly IIH, TOS CVH (!) and craniovenous outflow obstruction) will frequently cause severe fatigue, migraine, headache, dizziness, tinnitus, pain in the upper neck/back of the head (this is hypertensive migraine, not atlas pain — Larsen et al 2020), POTS, memory loss, cognitive decline or fluctuating cognitive ability, syncopal event, seizures, and even, sometimes, hemi or paraparesis and other stroke-like symptoms. These problems are much more constant than AAI CCI, which are, for the most part, positional problems. Moreover, craniovascular disorders often fluctuate depending on whether or not the patient is upright or lying down (sometimes lying down is worse, sometimes standing up makes it worse), and do certainly not return to normal, symptom-free status when the neck is placed in neutral position. Most imaging is tends to be normal, except certain craniovascular workups, especially a CTV of the head, TOS workups, and doppler of the carotid and vertebral arteries (not positive for hypoperfusion, but hyperperfusion). You can read more about these problems in my Myalgic encepalitis (link) and intracranial hypertension (linked earlier) articles as well as my 2018 and 2020 papers (Larsen 2018, Larsen et al 2020) in the reference lists if you think this may be you.
Case report
A 32 year-old female patient contacted me in 2019 as she had been diagnosed (by a radiologist alone) with craniocervical and atlantoaxial instability. The report claimed that there were signs of ligamentous rupture and bidirectional subluxation upon rotation in the atlantoaxial joints. She was also said to have ventral brainstem compression, which particularly scared her due to her difficulties with respiration. She was never evaluated for clinical correlation for these alleged findings, ie., no one evaluated if these findings had actual compatibility with her clinical symptoms and, especially, triggers.
The patient had headache, dizziness, fatigue, pain in the arms and chest and often felt difficulty breathing. She worsened with arm-loading, and often worsened when lying down, especially the breathing dysfunction tended to exacerbate and become more pronouned at night-time, resulting in anxiety and insomnia. She had been out from work for one year at the point of consultation, but her doctors could not find anything wrong with her. She started researching on certain online forums, in which she was advised to look into AAI and CCI. This, again, prompted the more than 1000 euro consultation with the upright imaging center in a large european country.
I consulted with her and reviewed her imaging: The quality of the images, first and foremost, was very low. The CXA was 138 degrees and the Grabb-Oakes measurement was 8,3mm. The BDI was 6mm and the BAI was 8mm, which are all farily normal. And, although there was zero evidence of brainsstem compression, she did indeed have subluxation of atlantoaxial joints with around 10% of overlap when turning to the side. Her symptoms, however, did not at all change when changing her neck position and she had never had torticollis. Rather, she would feel awful in general and felt worsening with stress and arm- & shoulder loading, and being upright vs. lying down. These are typical signs of craniovasculo-hypertensive disorders. All conventional things like heart and lung problems, MS, cancer, infections etc. had been excluded by her primary care physicians and local hospital. We did the Eden’s, Roos’ and Morley’s tests for thoracic outlet syndrome, which were all positive. We moved on to perform the Valsalva maneuver (a pressure test), the Queckenstedt’s test (manual venous compression test), and the cervical retraction test (TOS CVH), in which the first and third tests were positive, reproducing severe head pressure, dizziness, presyncope and profound fatigue. None of these tests would be able to reproduce her symptoms if they were stemming from AAI or CCI.
I diagnosed her with mild (“benign”) atlantoaxial instability and TOS CVH. I told her that, although I don’t think there’s any evidence to suggests that the AAI is causing your symptoms, we should still treat it to prevent the risk of future frank luxations of the joints. In addition to that we would start treatment for thoracic outlet syndrome. TOS is often considered a mere upper limb nerve pathology, but this is not the case. TOS increases perfusion rates to the brain, to which the brain is very sensitive and may dysfunction depending on how high the pressures are (Larsen et al 2020), often resulting in severe fatigue, dizziness, headaches and especially occipital headaches/pain (these are hypertensive headaches, not an atlas problem). TOS is also a common cause of dyspnea (respiratory difficulty), although these patients will have normal blood oxygen levels, which was also the case here. This, as significant irritation of the brachial plexus can also cause autonomic coaffection (Larsen et al 2021) and thus derange the function of the phrenic nerves, which in turn control the diaphragm.
From the beginning, the patient doubted my diagnosis that this was a craniovascular problem because she felt pain in the suboccipital area, had cracking and clunking, and felt compatible with several things she had read online and on facebook forums. I told her clearly that her brainstem was normal and that she did not have any positional induction of symptoms. Unfortunately, she was not compliant to the treatment that I prescribed (TOS, TOS CVH) other than the treatment for AAI, which she was convinced that was her problem. And, fair enough, I do not expect blind trust nor compliance. What’s interesting, regardless, is that one year after we had the first consultation she underwent another uMRI (due to lack of improvement of symptoms), which showed complete resolution of the atlantoaxial subluxations, where the facet joints were now overlapping at about 30%; 300% improvement (remember: >20% is normal). And, she still had the same symptoms! No improvement! This is what I said from the beginning; AAI is not the cause of these symptoms, the exam and triggers do not fit.
Case report #2
A 24 year-old male patient contacted me from various issues all over his body, randing from fatigue, dizziness, tinnitus, facial and jaw pain, low back pain, leg pain etc. He had been diagnosed with Chiari malformation, but told that it was early stage and non-sinister in nature, at least at the moment.
This is a case where a combination of mild craniocervical instability and congenital spinal canal hypoplasia causes the potential for misinterpretation of the Chiari malformation and its severity, because Chiari malformation should not be determined merely based on how far the cerebellar tonsils surpass McRae’s line, which in this case is “mere” 6mm. 6mm of descent is not normal, but in a patient with a normal craniocervical junction and normal spinal canal caliber, there will not be tight conditions for the cerebellar tonsils and therefore also no significant risk of developing syringomyelia or medullopathy. It is also worth noting that Harris measurement (BAI) is 13mm here, which is clearly abnormal, although there are no current clinical symptoms of brainstem injury. A CSF study was done, showing weak, but somewhat patent CSF flow through teh foramen magnum, which is why there is no syrinx in development at the point of the study.


I am not convinced that the patient’s symptoms are stemming from CCI, not at all. The brain MRI showed dilation of the lateral and third ventricles in the brain, and he had jugular vein stenosis as well. He said that both arms went numb when raising them, and also had all kinds of other classic TOS signs that would explain his neck, upper back and arm symptoms. His slump test was negative, and there is no extremity weakness. I diagnosed him with LPES for the lower body symptoms.
So, what is the point here, if CCI is not the culprit for his symptoms?
The point is that there is genuine future risk of cerebellar and possibly medullary injury if the patient remains undiagnosed and untreated. Note how, although there is “only 6mm” of descent, the posteriorly deflected medulla also pushes the cerebellar tonsils into the opisthion (the posterior rim of the foramen magnum) and therefore increases the risk of real compressive injury. He does probably also suffer from some kind of underlying hypermobility disorder such as Ehler Danlos or Marfan’s syndrome. This, in combination with poor posture and weak cervical musculature, over years, is most likely what has lead to the posterior migration of the odontoid process and thus an expansion of BAI. The patient was told that there was no need for concern and no need for further followup. This is false, and potentially dangerous advice. This, contrary to what he was told, is a case where mild and subclincial craniocervical instability combined with a narrow spinal canal and mild descent of the cerebellar tonsils, in tandem, has led to a potentially dangerous condition. I recommended a new head or neck MRI, flexion extension imaging, and referral to a new neurosurgeon with experience in the treatment and diagnosis of Chiari malformation for a second-opinion. It is not unlikely that this patient will require both occipital decompression as well as atlantoaxial fixation in the future.
Summary
Atlantoaxial and craniocervical instability are both real and potentially sinister diagnoses that require treatment. However, appropriate inclusive criteria must be used to render the diagnoses; subtle findings and the lack of a strong clinical correlation is not enough, and will easily lead to misdiagnosis and related anxiety and suffering. AAI and CCI are diagnoses that mainly cause the risk for either brainstem damage or injury to the arteries that supply the brain with blood, and this can cause paralysis or stroke if left untreated in cases where there is legitimate evidence for pathology. The other side of the AAI/CCI coin is the risk for facetal luxation; a less sinister-, but still a problem that warrants surgical treatment.
Important takeaways:
- Patients with legitimate CCI or AAI will generally have intermittent induction of symptoms with full rotation, flexion or extension that resolves in netural position, presuming there is no constant crushing of the brainstem or vertebral artery dissection
- Merely feeling worse when standing up, even if indeed feeling awful, is not a strong indicator of AAI CCI — As mentioned above, it is the influence of cervical positioning. Dysautonomia when standing up is often related to craniovascular problems, whereas difficulty holding the head up suggests mumscular damage.
- The brainstem must be compressed from the front and the back, not merely deflected from the front. Moreover, genuine cases of brainstem compression causes paralysis and other upper motor neuron signs, and will present with syringobulbia or compressive bulbopathy. Lack of signal change in the cord, and especially when it is not being compressed from both sides, is not a case of brainstem compression
- Mild to moderate ligamentous compromise in cases where all measurements are normal or nearly normal, and there is no neurovascular compression, is generally NOT a surgical indication nor an indication for aggressive treatment. Moreover, it would certainly not suggest a sinister future deterioration in the vast majority of circumstances
- Mild and often even moderate circumstances of AAI and CCI can be treated with appropriate (specific, not generic) physical therapy to strengthen the muscles that prevent hypermobility.
- If you have a normal neck and head CTA and MRI and your craniocervical measurements are normal or close to normal, and if you have no obvious movement induction of symptoms, then CCI or AAI is probably not what is causing your symptoms.
Be sure to understand the mechanism of induction of symptoms in AAI and CCI before jumping on this potentially dangerous, and often financially devastating bandwagon!
References
- Yang SY, Boniello AJ, Poorman CE, Chang AL, Wang S, Passias PG. A review of the diagnosis and treatment of atlantoaxial dislocations. Global Spine J. 2014 Aug;4(3):197-210. doi: 10.1055/s-0034-1376371. Epub 2014 May 22. PMID: 25083363; PMCID: PMC4111952.
- Fielding JW, Hawkins RJ. Atlanto-axial rotatory fixation. (Fixed rotatory subluxation of the atlanto-axial joint). J Bone Joint Surg Am. 1977;59 (1): 37-44. J Bone Joint Surg Am
- Spinnato P, Zarantonello P, Guerri S, Barakat M, Carpenzano M, Vara G, Bartoloni A, Gasbarrini A, Molinari M, Tedesco G. Atlantoaxial rotatory subluxation/fixation and Grisel’s syndrome in children: clinical and radiological prognostic factors. Eur J Pediatr. 2021 Feb;180(2):441-447. doi: 10.1007/s00431-020-03836-9. Epub 2020 Oct 16. PMID: 33064218.
- Ross & Moore. Diagnostic imaging: Spine, 3rd edition. 2015. Elsevier Publishing
- Deliganis AV, Baxter AB, Hanson JA, et al. Radiologic spectrum of craniocervical distraction injuries. Radiographics 2000;20:S237-50.
- Goel A. Facetal alignment: Basis of an alternative Goel’s classification of basilar invagination. J Craniovertebr Junction Spine. 2014 Apr;5(2):59-64. doi: 10.4103/0974-8237.139199. PMID: 25210334; PMCID: PMC4158632.
- Burry HC, Tweed JM, Robinson RG, Howes R. Lateral subluxation of the atlanto-axial joint in rheumatoid arthritis. Ann Rheum Dis. 1978 Dec;37(6):525-8. doi: 10.1136/ard.37.6.525. PMID: 749697; PMCID: PMC1000289.
- Musa A, Farhan SA, Lee YP, Uribe B, Kiester PD. Traumatic Atlantoaxial Lateral Subluxation With Chronic Type II Odontoid Fracture: A Case Report. Int J Spine Surg. 2019 Feb 22;13(1):79-83. doi: 10.14444/6010. PMID: 30805289; PMCID: PMC6383461.
- Ujifuku K, Hayashi K, Tsunoda K, Kitagawa N, Hayashi T, Suyama K, Nagata I. Positional vertebral artery compression and vertebrobasilar insufficiency due to a herniated cervical disc. J Neurosurg Spine. 2009 Sep;11(3):326-9. doi: 10.3171/2009.4.SPINE08689. PMID: 19769514.
- Josy GF, Daily AT. Bow hunter’s syndrome revisited: 2 new cases and literature review of 124 cases. J NS 2015, V8 issue 4. E7. DOI: 10.3171/2015.1.FOCUS14791.
- Faris AA, Poser CM, Wilmore DW, et al.. Radiologic visualization of neck vessels in healthy men. Neurology. 1963;13(5):386–396.
- De Kleyn A, Nieuwenhuyse P. Schwindelanfalle und Nystagmus bei einer bestimmten Stellung des Kopfes. Acta Otolaryngol. 1927;11(1):155–157.
- Henderson FC Sr, Rosenbaum R, Narayanan M, Koby M, Tuchman K, Rowe PC, Francomano C. Atlanto-axial rotary instability (Fielding type 1): characteristic clinical and radiological findings, and treatment outcomes following alignment, fusion, and stabilization. Neurosurg Rev. 2021 Jun;44(3):1553-1568. doi: 10.1007/s10143-020-01345-9. Epub 2020 Jul 4. PMID: 32623537; PMCID: PMC8121728.
- Myran R, Kvistad KA, Nygaard OP, Andresen H, Folvik M, Zwart JA. Magnetic resonance imaging assessment of the alar ligaments in whiplash injuries: a case-control study. Spine (Phila Pa 1976). 2008 Aug 15;33(18):2012-6. doi: 10.1097/BRS.0b013e31817bb0bd. PMID: 18708935.
- Gweon HM, Chung TS, Suh SH. Evaluation of the Cause of Internal Jugular Vein Obstruction on Head and Neck Contrast Enhanced 3D MR Angiography Using Contrast Enhanced Computed Tomography. J Korean Soc Magn Reson Med. 2011 Apr;15(1):41-47. English. https://doi.org/10.13104/jksmrm.2011.15.1.41
- Dashti SR, Nakaji P, Hu YC, Frei DF, Abla AA, Yao T, et al. Styloidogenic jugular venous compression syndrome: diagnosis and treatment: case report. Neurosurgery. 2012 Mar;70(3):E795-9. doi: 10.1227/NEU.0b013e3182333859.
- Larsen K. Occult intracranial hypertension as a sequela of biomechanical internal jugular vein stenosis: A case report. Anaesth Pain & Intensive Care 2018;22(2):238-242
- Higgins N, Pickard J, Lever A. Lumbar puncture, chronic fatigue syndrome and idiopathic intracranial hypertension: a cross-sectional study. JRSM Short Rep. 2013 Nov 21;4(12):2042533313507920. doi: 10.1177/2042533313507920. PMID: 24475346; PMCID: PMC3899735.
- Li M, Gao X, Rajah GB, Liang J, Chen J, Yan F, et al. Styloidectomy and Venous Stenting for Treatment of Styloid-Induced Internal Jugular Vein Stenosis: A Case Report and Literature Review. World Neurosurg. 2019 Oct;130:129-132. doi: 10.1016/j.wneu.2019.06.100. Epub 2019 Jun 21.
- Özen Ö, Ünal Ö, Avcu S. Flow volumes of internal jugular veins are significantly reduced in patients with cerebral venous sinus thrombosis. Curr Neurovasc Res. 2014 Feb;11(1):75-82. ncbi.nlm.nih.gov/pubmed/24321024
- Higgins JN et al. Headache, cerebrospinal fluid leaks, and pseudomeningoceles after resection of vestibular schwannomas: efficacy of venous sinus stenting suggests cranial venous outflow compromise as a unifying pathophysiological mechanism. J Neurol Surg B. DOI: 10.1055/s-0039-1677706
- Perez MA, Bialer OY, Bruce BB, Newman NJ, Biousse V. Primary Spontaneous Cerebrospinal Fluid Leaks andIdiopathic Intracranial Hypertension. Journal of Neuro-Ophthalmology 2013;33:330–337doi: 10.1097/WNO.0b013e318299c292
- Alkhotani A. Cerebrospinal Fluid Rhinorrhea Secondary to Idiopathic Intracranial Hypertension. Case Rep Neurol 2019;11:295–298 https://doi.org/10.1159/000503813
- Waldock WJ, Higgins NJ, Axon P. A case report of gastroparesis resolved by styloidectomy. Otolaryngology Case Reports Volume 16, September 2020, 100201
- Larsen K, Galluccio FC, Chand SK. Does thoracic outlet syndrome cause cerebrovascular hyperperfusion? Diagnostic markers for occult craniovascular congestion. Anaesth pain intensive care 2020;24(1)69-86. DOI: https://doi.org/10.35975/apic.v24i1.1230
Super interesting article!
Would this mean that upper cervical chiropractors (orthogonal, blair technique, gonstead, etc.) are generally useless in most cases?
Why do they have results tho’ when they correct the atlas/axis?
How is possible for them to have results when there is no symptomatic AAI/CCI?
Maybe they temporary fix some compression?
Tks,
1. I believe that most of these practitioners mean well. That said, yes, it is my opinion that the treatment is nonsense.
2. My experience has been that these approaches do not work, and certainly do not cause long term results. I, personally, although I created my own manipulation protocol for this problem — ALMOST NEVER use it. Because it doesn’t work most of the time, and doesn’t cause any lasting results. The alignment of the atlas itself isn’t really the problem; the problem is whether or not a rotation or a horizontal glide is causing encroachment of the jugular outlet. If a gliding is causing it (it is usually a glide or, a glide combined with mild rotation), no manipulation can fix it. If it’s caused by rotation (rare), manipulation may temporarily improve jugular outlet passage, but it will not last. Moreover, tractioning the neck of these vulnerable patients can often cause undesirable effects.
3. Not sure what you mean here. If there are no symptoms, then what reuslts are you talking about? Or do you mean that there are positive improvement in symptoms despite the imaging being labeled as negative? If the latter, could be JOS obstruction, or could be placebo. A lot of things that cause temporary results are just placebo.
4. I already answered this.
If someone has an ADI of 4.5mm, can this be treated via physical therapy, or is it too much instability? What muscles would need to be strengthened to prevent the ADI from opening up?
Would need a flexion extension MRI and correlate to the patient’s symptoms. Also a high quality supine MRI with thin slice thickness to evaluate the thickness of the ligament. ADI laxity is mainly caused by head and neck trauma, so as long as you avoid future collisions, it will probably not deteriorate. Having a strong neck and good posture helps a lot as well (details on what this entails can be read in my article on atlas instability)
You mention to test for craniovascular pathologies, we should get a Doppler examination of the carotid and cerebral arteries done, and a CT angiogram done.
Does it matter whether these are done laying or sitting down? My symptoms are mostly sitting or standing but better laying down, won’t doing the CT angiogram then become useless if I do it laying down (my symptoms are dysautonomia-like when standing)
The exam should be done lying down, without a neck pillow. It is possible to do it with extension and rotation, etc., but it is usually not necessary. Exam for bow hunter’s syndrome is done dynamically, but that’s aother exam.
I received a Chiari Decompression two months ago after complaints of persistent dizziness with head movement. It was a craniectomy and C1 laminectomy with no dural opening. My chief complaints were a dizziness anytime I moved my head up and down or left and right (kind of like feeling on a boat) and numbness and tingling radiating from my shoulders down to my hands on both sides.
My clivo axial was 138 and my grab oakes was 8mm. My neurosurgeon still thought that the surgery would relieve my dizziness and symptoms given the cerebellar compression, but I am worse off since the procedure. My dizziness is more constant now and my numbness and tingling is still there.
I’m working with a PT to address my symptoms, mostly through vestibular therapy and postural exercises, but now I’m getting more numbness radiating from my shoulders down when doing the exercises, and am also getting more dizzy when I do the VT exercises (like neck movement left right up and down in quick motion), but not getting any better.
Do you have any recommendations? My PT thinks I have TOS and upper cervical instability based on my symptoms from the exercises, but doesn’t have any insight into what to do to treat either of them without looking things up on the internet herself. Based on your article, I would tend to agree with her assessment. But I don’t know how to tackle the dizziness or posture without bothering one another’s symptoms.
Feeling very lost…
I’d have to review your case in its entirety and put you through various tests.
I’m commenting here with the off chance I can solve the riddle of my head and neck pain, neurological symptoms or prevent further harm to my spine and brain. July 2019 ACDF C5-6-7, Feb 2020 right vertebral dissection leading to the diagnosis of Fibromuscular Displaysia, March 2020 another dissection of right vertebral artery, pseudo aneurysm of carotid artery and 2 in basilar artery. April 2022 learned my spinal fusion had failed at the level of C6-7, as I sought a 2nd opinion for surgery May 2020 I next found myself in the Emergency Room with a carotid dissection at the level of C1 limiting blood flow significantly resulting in stroke, I received 2 stents in tandem. I just went in to my former spine surgeon for new MRI imaging Feb 2023 and currently only have radiology notes to go by as he has dismissed me as a patient, I am currently waiting for a transfer of records and another 2nd opinion with a neurosurgeon. Latest MRI states disc herniating C4, slight curvature to left, anterolisthesis, and arthropathy. My original MRI 2019 pre surgery did note a small cerebellar tonsillar ectopia and later my concern was a family history of Chiari. I feel like I don’t even know where to look for answers, I have been doing PT and OT for 3 months now without success. Headache, neck pain, nerve pain, pain in back shoulder area and chest, gagging, insomnia, involuntary limb movement, tinnitus, numbness and weakness in my hands and 1 foot, fatigue, vision loss, nystagmus, severe dizziness, all for the last 4 years is wearing thin for my patience and I’m sure that of my GP. I’ve tried too many drugs to count and I want my life back before there isn’t one left. Any suggestions would help, I understand your warning of patients and overzealous diagnosis but I feel like a victim of negligence as well as discrimination, and whatever code that keeps doctors protecting other doctors at the cost of the patient.
Kim
What you’re describing here (cervical hernias and a carotid dissection) are not indicative of AAI nor CCI. The symptoms that you list would need clinical correlation to fit with those diagnoses, and that’s not something one merely picks off a symptom chart (I am not saying you’re doing that, but many are, and they end up paying for it).
There could be that your imaging or other unmentioned information, is, however, but based on what you write here, my first go-to would be a CTA of the brain and neck along with a new MRI of the neck. With regards to discrimination, I wouldn’t be able to have an opinion on that, but negligence is certainly possible if you have failing hardware after neck surgery and possibly additional untreated severe sites of nerve root compression.
There’s no way to give you any straight answer on this without a through review of your case.
Whats the atlas Larsen so I can buy it?
No idea what you’re asking here
Very fortunate to come across this article. I was fairly convinced that I’ve been suffering from mild CCI but haven’t had a formal diagnosis yet. I’ve been seen by multiple doctors and currently do PT, Rolfing, Massage, and Chiropractic to manage my symptoms, which for the last year has mostly been dull pain in my cervical spine. Quick summary of events:
July 2021 – Began feeling pain in my neck and base of skull. Dismissed as minor tension headache which persisted daily.
August 2021 – Fainted twice on plane after watching violent movie (seeing violence and blood never bothered me before)
Sept 2021 – Fainted again on return plane ride home after trying to watch the same movie because I couldn’t believe what happened.
Oct 2021 – After yet another trip, I woke up the next morning to my head pressured up, mind racing, vertigo, dizziness, anxiety, and pain in the neck. Also developed pulsatile tinnitus which would disappear when moving my neck the right way. This persisted for quite a while and even to this day I still occasionally get it.
Sought chiropractic care and massage therapy. After 3 weeks, pressure subsided, but other symptoms took a bit longer to subside. Eventually left feeling pain daily in the cervical spine which sometimes radiated to the back of the head and down between the shoulder blades.
I’ve had an MRI of the head/brain with and w/o contrast – All doctors reported as being normal
MRI of the cervical spine with and w/o contrast – Again, not much noted by most doctors, but an orothopedic interventionalist in Colorado who runs a specialty clinic noted I have mild cervical spondylosis and perhaps minor CCI
Ultrasound of Carotid artery was normal
Finally I also had a DMX done with this impression (I appreciate your skepticism of these):
• Damage to the posterior longitudinal ligament is indicated by an anterolisthesis at C5 on C6. (1mm C4-C5, 2.2mm C5-C6)
• Damage to the anterior longitudinal ligament is indicated by a retrolisthesis at C4 on C5 and C5 on C6 and anterior widening of the intervertebral disc space at C4-C5 and C5-C6. (2.2mm C4-C5, and 2.7 mm C5-C6)
• Damage to the alar and accessory ligaments is indicated by an overhang of the lateral mass of C1 bilaterally. Also change in the para-odontoid space during bilateral lateral bending. (3.9 mm head tilted to the right, 3.7 mm head tilted to the left)
Even regular x-rays where I tried to hold my head and neck in different directions seemed unremarkable.
Fast forward to yesterday, I had a snycopal episode at work after not having one since Sept ’21 also felt a little bit of tingling in the left arm before it happened and after. I’m seeing another doctor and getting an echocardiogram and EKG to examine my heart.
After finding this article, I was wondering if you might seem to think this is actually TOS CVH and whether I should pursue the other testing for it? My pain persists daily, but I usually feel the best in the morning after sleeping and resting all night… within an hour or two, my neck begins to feel tight and low grade aching pain persists. I’m just slightly worried about the syncopal episodes as I work in a dangerous environment and wonder what sort of treatment might be suggested if it was indeed TOS CVH? Thank you Kjetil.
Would vascular TOS cause increase of dizziness when looking down or up? Or it’s more likely that arteries are compressed by CCI/AAI
Sounds vestibular. Vestibular problems often have vascular underlying causes.
Dear Kjetil, your work on the issue is refreshing. I learnt so much about the mechanisms affecting/causing CCI and will be following your moves. I really like the way you are approaching the problem – we are in the end a bio machine and need a good bio engineer to diagnose us. Where can I find the videos? Can I buy them/subscribe?
Dear Kjetil, your work is refreshing and I find it inspirational. I learnt so much from your articles and videos and realised that what my old physiotherapist back in Poland was telling me for years was right: strengthening the muscles is probably the paramount part on the road to recovery and maintaining rehab results. I was mad into stretches for years doing Pilates and all sorts of dynamic stretches. However, when my neck started to cause serious troubles lately (most probably due to the shock after manual therapy/neck adjustment) I foolishly started to stretch like crazy all my neck muscles that are probably very stretched at that stage due to the previous exercise. I developed monstrous headaches that incapacitated me for two weeks and then fluctuated. Tinnitus, headache, insomnia, dizziness, you call it. I was terrified that my problems might have been ruptured or dissected vertebral arteries (my mum has a history of cranial hypertension and mini strokes) so I asked for an MRI which showed no damage to the muscles, ligaments or veins. After watching and reading all your work I decided to talk to ask her to read your work (hope she is open minded:-) and will send my scans to you for a review in accordance with service offer. Thank you again.
Could an upright cervical MRI in flexion, extension and maximal bi-directional rotation also show signs of craniovenous hypertension and TOS CVH? And which tests would you recommend for someone who thinks they have CCI but is more probablycraniovenous hypertension and/or TOS CVH who cant afford doing all of the listed ones in this paper?
These are different things. Read the article, I show my recommendations there.
I have a report with images and measurements that states I have CCI and would benefit from tx with prolotherapy. Could I send it to you and you tell me if my diagnosis is legit or if it’s actually not that bad, and I’m falling victim to a scam? Thanks
Yes
This post has been so helpful thank you. MY DMX rays from June revealed I have anterolisthesis at C3-C4 and C4-C5 (2.4mm and 2.2mm respectively) as well as retrolisthesis at the same areas (2.3mm and 2.5mm). I’m concerned most with my overhang of lateral mass of C1 to left (2.7mm). Dizziness, headaches, ear pressure, bobbling head and heart flutters, etc. I received PRP to the functional Spinal unit everywhere but C1-C2. Based on those measurements, is this a pretty average case? Or do you think it’s more severe? I have all the imaging if you’re able to look and give your 2 cents. Thanks for your work on this
Can’t comment on cases like this without seeing the pictures, but a 2,7mm lateral overhang wouldn’t worry me much at all.
For those with moderate cci/AAI, are the neck isometrics contractions the only exercise possible, since moving the neck with range of motion exercises would cause flareup and possible further damage?
If you literally can’t move your neck without putting yourself in danger, you need surgery. That said, I doubt that’s the case; so many tell me similar things and have absolutely normal or virtually normal imaging.
Thank you for this article. Im dealing with alot of these symptoms, including some that are a bit extreme like drenching sweats when I move my neck or for some reason when i need to pee. I can get my symptoms under control laying down on my side (cant take any pressure even pillows so no back sleeping), or wearing a brace, but my lordosis means a brace is trying to force a curve and it strains my neck muscles. Interestingly pain was not a major symptom for me until recently. It started with central apnea, crazy sweats, and hearing loss/ Tinitus, general weakness. Oh and intermittent violent vertigo, with head turning. And when I say violent I mean VIOLENT but also very intermittent meaning i cant reproduce it and its not frequent at all. Symptoms worse in extension and when I straighten posture and often when i move at all tbh. If only I could sit perfectly still for the rest of my life.
My mri from 2019 looks ALOT like the one you have of the person who is emerging cci likely with underlying EDS or Marfans. Tipped odinoid, larger basion measurements, grab oakes around 11mm, chiari around 4 mm with very crowded tonsils. My CxA is around 130, so not horrible. Guess I will wait and see how my mri looks next week, because whatever is happening, I cant live this way anymore.
You are definitely right that the problem is that local Drs basically throw their hands in the air, have no differential diagnosis, no other possibilities, and often act like its in your head. We are then forced to go to the only Drs aware that this is a possibility with EDS.
Thank you for being the flip side of the coin. I wish I could see one of the main Drs AND a Dr who is knowledgeable yet conservative. Thats ideal for ALL medicine.
Hello. I stumbled upon CCI after I had a recent flex/ext upright cervical MRI with a 133 CXA neutral, 127 flexion, 149 ext. I had asked for this from a neurosurgeon at Johns Hopkins because my chiropractor and I had realized that my symptoms (leg weakness and other neurological symptoms) would be alleviated/exacerbated with different positions of my neck. The cervical chiropractic adjustments would completely alleviate my symptoms for a few hours, but then reverted right back with no long term improvement. I found out about CCI after googled the CXA finding on my MRI. I recently tried out a cervical collar and it also alleviates my symptoms (entirely or close to it). The neurosurgeon at JH had never heard of CCI and told me to keep looking for an answer. Others have been much less supportive. Anyway, I don’t have a diagnosis, but am wondering if it would be worthwhile to wear the cervical collar until I get this figured out. I am 100% reliant on crutches without it – and surprisingly just took my first walk around the neighborhood in almost 4 months with the cervical collar.
Hi Carrie
It really depends on what degree of leg weakness we are talking about. CCI would only cause leg weakness if it is seriously impinging the spinal cord, which is very unlikely and would be easily diagnosed on a flexion / extension as an emergency. The other possibility is vertebral artery compromise, but I don’t think it fits well with what you are describing. Either way, to give any conclusive answers I would have to review your case myself.
Best wishes.
Hello. This article is very helpful in looking for actual problems. 2 1/2 years ago i started to have neck pain because of falling from bike. For which i started to stretch my neck in flexion, extension, side bending and rotating directions which helped reduce pain for some while. But one day when doing this i unfortunately stretched very strong i guess which lead to hypermobile, head dropping in my head. Within few hours started to feel numbness in left side body. Went neuro and took mri for brain and neck which came back normal. The head dropping sensation reduced i believe bcs of muscle tightened up .the numbness went but i started to feel weakness in left and right side body with left being more prominent and no notable neurological deficts.I also started to have floaters and visual snow which the eye doctors said nothing wrong with my eyes Over the course of 2 years i went to 4 to 5 neurosurgeon who ordered flexion extension x ray of neck, mri of neck and brain which all came back normal. But for the past one month the loose feeling in neck came back with more severe weakness in body which increases even more if i m upright fir little long. I went to neuro who asked mra of neck and brain which came back normal. This symptoms does nt increase or reduce in any position of neck and there constantly. I m completely lost as there is no way infront for recovery with increasing weakness. Is there any thing i can do to diagnose my issues
Most likely you have stretched your neck so much that you made the initial damage much worse. Very specific rehabilitation of the neck will be required to recapacitate the cervical musculature.
Hello,
Thank you for the article.
Unfortunately, I mimic the 32 yo female patients symptoms from your case study (I’m 36 female but my symptoms started when I was 25) who you suspected had tos cvh cranial venous outflow obstruction.
I did do genetic testing to confirm KEDS, and have had several doctors confirm I am hyper-mobile.
My husband and I have been trying for years to have me properly diagnosed and treated. We have been going down the cervical instability path for a few years now.
We were wondering what the recommended treatment plan was for the patient (even though she declined it)? We are going to request the tests to be ordered for myself.
Thank you!